What’s your QT Interval?
What is the QT interval? From the beginning
of the Q to the end of the T is the QT interval.
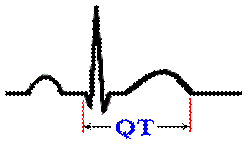
What
does the QT represent? The QT interval measures the
complete ventricular contraction and relaxation cycle.
Why
do we have to look for the QT Interval?
The
longer the QT interval the higher risk of lethal cardiac arrhythmias,
specifically Polymorphic Ventricular Tachycardia known at “Torsades de Points”.
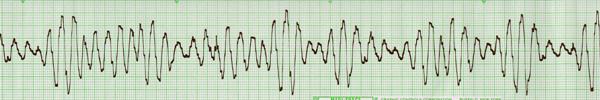
Torsades de
Pointes
Thousands
(5,000 -7,000) of apparently health children and adults die annually due to
this under recognized condition.
What
are the symptoms? Sudden loss of consciousness (the
medical term is 'syncope') and sudden death are the common symptoms and usually
occur during physical exertion or emotional excitement like anger, fear or
startle, but may occur during sleep, or arousal from sleep. Common startle
events include sirens, the telephone and the alarm clock. It is less common for
the syncope or sudden death to occur when the person is awake and at rest. The
particular trigger for the symptoms depends to some degree on the specific gene
abnormality (see the section on genetics). Exercise induced syncope usually
occurs right during the exercise, but occasionally occurs within a few seconds
or a minute or two after the exertion. In patients who experience syncope the
torsade de pointes rhythm reverts spontaneously to normal, usually within about
1 minute or less. When this occurs, the patient quickly regains consciousness,
usually without disorientation or residual symptoms, although fatigue may be
present. When the torsade rhythm persists for a longer time, however, it
degenerates into ventricular fibrillation and the outcome is death unless
electrical defibrillation is provided.
Not all patients who have this condition
have symptoms; at least one-third, and probably more, never develops any
symptoms. In the others, some have just one or two syncopal spells as children,
and none thereafter, whereas, some have many episodes over a number of years.
The symptoms may begin as early as the first days or weeks of life, or as late
as middle age. Most commonly, however, the symptoms first occur during pre-teen
and teenage years. The symptoms start earlier in males than females, beginning
on average at approximately 8 years in males and 14 years in females. Because
many affected persons never have symptoms, the absence of a history of syncope
or sudden death in a family does not at all guarantee the absence of LQTS in
the family.
How
do you get Long QT?
Ionic
Channelopathies. “A dysfunction of
special heart cells called ion channels” These channels control the flow of
ions like potassium, sodium and calcium molecules in and out of the heart
cells. This flow produces the electrical activity of the heart.
You can
generally get these in two ways.
1. Inherited from genetics, e.g.
passed on from mom or dad to
child.
or
2. Acquired through medication use.
There are 8 LQT genotypes that
have been identified so far.
LQT1
LQT1 is the most common type of long QT syndrome, making
up about 40 to 55 percent of all cases. The LQT1 gene is KCNQ1 which has been
isolated to chromosome 11p15.5. KCNQ1 codes for the voltage-gated potassium
channel KvLQT1 that is highly expressed in the heart. It is believed that the
product of the KCNQ1 gene produces an alpha subunit that interacts with other
proteins (particularly the minK beta subunit) to create the IKs ion
channel, which is responsible for the delayed potassium rectifier current of
the cardiac action potential.
Mutations to the KCNQ1 gene can be inherited in an autosomal dominant or an autosomal recessive pattern in the same family.
In the autosomal recessive mutation of this gene, homozygous
mutations in KVLQT1 leads to severe prolongation of the QT interval (due to
near-complete loss of the IKs ion channel), and is associated with
increased risk of ventricular arrhythmias and congenital deafness. This variant
of LQT1 is known as the Jervell and Lange-Nielsen syndrome.
Most individuals with LQT1 show paradoxical prolongation
of the QT interval with infusion of epinephrine.
This can also unmark latent carriers of the LQT1 gene.
Many missense
mutations of the LQT1 gene have been identified. These are often associated
with a high risk percentage of symptomatic carriers and sudden death.
LQT2
The LQT2 type is the second most common gene location
that is affected in long QT syndrome, making up about 35 to 45 percent of all
cases. This form of long QT syndrome most likely involves mutations of the human
ether-a-go-go related gene (HERG) on chromosome 7. The HERG gene (also known
as KCNH2) is part of the rapid component of the potassium rectifying current (IKr).
(The IKr current is mainly responsible for the termination of the cardiac action potential, and therefore
the length of the QT interval.) The normally functioning HERG gene allows
protection against early after depolarizations (EADs).
Most drugs that cause long QT syndrome do so by blocking
the IKr current via the HERG gene. These include erythromycin,
terfenadine,
and ketoconazole.
The HERG channel is very sensitive to unintended drug binding due to two aromatic amino acids,
the tyrosine
at position 652 and the phenylalanine at position 656. These amino acid
residues are poised so drug binding to them will block the channel from
conducting current. Other potassium channels do not have these residues in
these positions and are therefore not as prone to blockage.
LQT3
The LQT3 type of long QT syndrome involves mutation of
the gene that encodes the alpha subunit of the Na+ ion
channel. This gene is located on chromosome 3p21-24, and is known as SCN5A
(also hH1 and NaV1.5). The mutations involved in LQT3 slow the
inactivation of the Na+ channel, resulting in prolongation of the Na+
influx during depolarization. Paradoxically, the mutant sodium channels
inactivate more quickly, and may open repetitively during the action potential.
A large number of mutations have been characterized as
leading to or predisposing LQT3. Calcium has been suggested as a regulator of
SCN5A, and the effects of calcium on SCN5A may begin to explain the mechanism
by which some these mutations cause LQT3.
LQT5
is an autosomal dominant relatively uncommon form of
LQTS. It involves mutations in the gene KCNE1 which encodes for the potassium
channel beta subunit MinK. In its rare homozygous forms it can lead to Jervell and Lange-Nielsen syndrome
LQT6
is an autosomal dominant relatively uncommon form of
LQTS. It involves mutations in the gene KCNE2 which encodes for the potassium
channel beta subunit MiRP1, constituting part of the IKr repolarizing
K+ current.
LQT7
Andersen-Tawil syndrome is an autosomal dominant form of LQTS associated with
skeletal deformities. It involves mutation in the gene KCNJ2 which encodes for
the potassium channel protein Kir 2.1. The syndrome is characterized by Long QT
syndrome with ventricular arrhythmias, periodic paralysis and skeletal
developmental abnormalities as clinodactyly, low-set ears and micrognathia.
The manifestations are highly variable.
LQT8
Timothy's syndrome is due to mutations in the calcium channel
Cav1.2 encoded by the gene CACNA1c. Since the Calcium channel Cav1.2 is
abundant in many tissues, patients with Timothy's syndrome have many clinical
manifestations including congenital heart disease, autism, syndactyly and
immune deficiency.
Associated
syndromes
A number of syndromes are associated with LQTS.
Jervell
and Lange-Nielsen syndrome
The Jervell and Lange-Nielsen syndrome
(JLNS) is an autosomal recessive form of LQTS with
associated congenital deafness. It is caused specifically by mutation of the
KCNE1 and KCNQ1 genes.
In untreated individuals with JLNS, about 50 percent die
by the age of 15 years due to ventricular arrhythmias.
Romano-Ward
syndrome
Romano-Ward syndrome is an autosomal dominant
form of LQTS that is not associated with deafness.
What
Drugs Prolong the
QT interval?
Please ask yourself, have I ever given these drugs to
my patient. These are very common
drugs.
These
drugs have a risk of Torsades de Pointes *,**
*As of
3/02/2006 ** From www.qtdrugs.org
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
Drugs that
prolong the QT interval and/or induce Torsades De Pointes |
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|
|
|
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
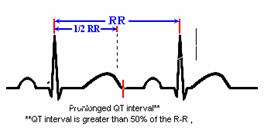
Is
the QT interval hard to Identify? No, it only takes
about 5 seconds to measure the QT interval. A simple QT measurement can help you recognize
the precursor to LQTS and prevent life threatening problems.
How
do you measure the QT interval?
The QT
interval should be less than half of the R-R interval. This is generally
accurate for regular rates between 65 and 90.
If you have a rate less than 65 and have a prolonged QT interval
consider a 12 lead ecg. This doesn’t
work on irregular rhythms.
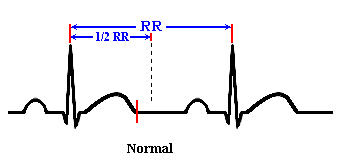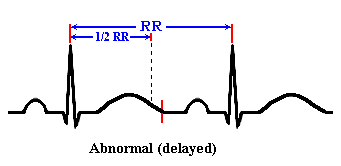
1)
Measure
the QT interval.
2)
Compare
the QT interval to the R to R interval
3)
If
the QT interval is greater than 50% of the R to R, it suggests LQTS.
4)
Notify
the MD in charge.
5)
Request
a 12 lead EKG to monitor for QTc > 440ms in men and >450/ms in women and
suggest cardiology screen for LQTS.
Long QT Work up
In 12%
of patients with LQTS, sudden death was the first manifestation of the disease
and in 4% this happened in the first year of life. This point alone
mandates
the treatment of all those diagnosed as affected, even if there are no
symptoms. In most cases, several members
of the same family are gene-carriers. Low penetrance exists in LQTS, which
means that gene-carriers may not show the clinical phenotype and may have a
normal QT interval. Therefore a normal QT in the parents does not rule out
familial LQTS. In addition, approximately 30% of cases are due to 'de novo'
mutations which imply unaffected parents and no family history. 'De novo' LQTS
mutations have been demonstrated in infant victims of cardiac arrest and sudden
death diagnosed as Sudden Infant Death Syndrome.
Even
though relatively few LQTS patients have cardiac events during the first year
of life, the vast majority become symptomatic later on, either during childhood
or adolescence according to genetic subgroups. Therefore treatment must
continue. Beta-blockers are the first choice therapy in LQTS and are effective
in preventing recurrences in 80% of already symptomatic
patients;
different degrees of protection exist according to genetic subgroups. If
beta-blockers are unable to prevent new cardiac events, additional drug
therapy, left cardiac sympathetic denervation, pacemakers or the implantable
cardioverter defibrillator should be considered based on evidence, with due
consideration for body size.
It is well understood that the likelihood of
having LQTS increases with increasing QTc; however, since a small percentage of
LQTS patients has a QTc <440 ms, the correlation between QT prolongation and
the presence of the syndrome is not absolute. Therefore, the following
discussion is presented as guidelines based upon experience and current
knowledge, and is likely to be updated frequently. Given the life-threatening
potential of the disease, once the diagnosis of LQTS becomes probable, it is
recommended that these infants are referred to a specialist as soon as
possible.
First ECG: QTc above 440 ms, the upper limit of normal.
Exclude
other causes of acquired QT interval prolongation and obtain a detailed family
history for the possibility of familial LQTS. Episodes of early sudden death,
fainting spells, and seizures epilepsy should alert to this possibility. The
ECG should be repeated after a few days to confirm the abnormal finding.
Subsequent management depends on
1.
presence or absence of family history suggestive for LQTS,
2.
the degree of QT interval prolongation.
The
presence of complex ventricular arrhythmias would have additional importance.
The following stepwise approach involves infants with and without a family
history for LQTS (see Figure 4 of the
original
guideline document).
If
family history is positive, then, as LQTS is an autosomal dominant disease, the
infant has a 50% probability of being affected and complete diagnostic
procedures should be performed, as always with LQTS families.
The second ECG is normal.
If the
first QTc was <470 ms, dismiss the case. If the first QTc was less than or
equal to 470 ms, then plan a third ECG after 1-2 months to remain on the safe
side.
The second ECG shows a QTc between
440 and 470 ms.
In these
cases with persistent borderline QT prolongation, electrolytes, including
calcium and magnesium, should be checked. Clinical history of autoimmune
disease and plasma titres of maternal antibodies (anti Ro/SSA and antiLa)
should be performed. T wave morphology may be
helpful;
for example, the presence of notches on the T wave in the precordial leads
further suggests the presence of LQTS. Additionally, mild bradycardia can also
be found in LQTS. ECGs should be obtained from the parents and siblings of the
neonate. In the absence of family history of LQTS, symptoms or arrhythmias, a
24-hour Holter monitoring should be obtained to look for T wave alternans,
complex ventricular arrhythmias or marked QTc prolongation, and the ECG should
be periodically checked during the first year. No treatment is currently
recommended. With a positive family history, the probability of LQTS becomes
high. Additional diagnostic procedures (24-hour Holter monitoring,
echocardiogram and genetic screening) should be performed and initiation of
therapy could be considered.
The second ECG shows a QTc greater
than or equal to 470 and <500 ms.
All
diagnostic procedures listed above should be performed and a third ECG should
be planned within a month. In case of a positive family history, therapy should
be initiated. Even without a family history, therapy should be considered. Even in infants with a very prolonged QTc in
the first month of life, the ECG may normalize. If subsequent ECGs and
diagnostic procedures do
not
confirm the presence of LQTS, it is logical to progressively withdraw therapy
and to return to periodic observations.
The second ECG shows a QTc greater
than or equal to 500 ms.
Infants
with a QTc >500 ms are very likely to be affected by LQTS and become
symptomatic. All diagnostic procedures listed above should be performed and
these infants should be treated.
Highest
risk. The
presence of QTc close to 600 ms, or of T wave alternans, or of 2:1 AV block
secondary to major QT prolongation, or of hearing loss, identify infants at
extremely high risk.
ST
segment elevation
Work-up:
Whenever the underlying cause has been identified, it should be
treated.
If the Brugada syndrome is suspected, careful family history should be
collected, 24-hour Holter monitoring obtained, and the patient should be
referred to a specialist.
Frequently asked Questions.
Q.
What does the QTc mean?
A. The QT interval corrected for the rate.
Q. Why does the QT interval need to be corrected for the rate?
A. The faster the rate the more
narrow the QT interval.
The slower the rate the wider the QT
interval.
The QT interval is not a static number that
we can tell you to look for.
Q. Where do you find the QTc?
A. The QTc is
calculated by all 12 lead EKG machines and is in the same area that contains
all other measurements on a 12 lead EKG. (e.g. Rate, axis, PR interval, QRS
interval etc… QTc is included in the
same area; just look for it and you will find it).
Q. What is normal
QTc?
A. Normal QTc; less
than 440ms in men
less than 450ms in women.
If your
pt has a prolonged QTc generally it’s a cardiology evaluation to rule out Long
QT syndrome. We don’t panic for QTc’s
that are a little long. The longer the
QTc gets the more concern we have.
Example;
A QTc of 580ms is definitely a cause for concern and needs a cardiology
evaluation along with a review of current medications. A QTc of 450ms is less of a concern but still
could be enough to consider a cardiology evaluation.
Q.
Do you need to document Qt interval and QTc?
A. Yes, and also document that you informed the
MD in charge of the pt’s care and pass it on to your co-workers in shift change
report.
Q. What formula is used to
calculate QTc?
A. Bazett's formula:
QT interval corrections in the literature use Bazett's
formula, defined as the observed QT interval divided by the square root of the
R-R interval in seconds. A corrected QT interval of > 440 ms is defined as generally
defined as abnormal. Bazett's formula corrects or normalizes the measured QT
interval for a heart rate of 60 BPM. Thus, the QT is measured at the given
heart rate, and the QTc estimates what the QT interval would be if the heart
rate were 60. Bazett's formula works reasonably well at "normal"
heart rates, but is less accurate when the heart rate is slow or fast.
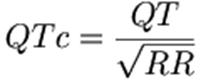
Rautaharju’s
formula:
A more accurate
method to correct the QT interval for the rate was developed by Rautaharju et
al., who developed the formula. This method is not widely used by clinicians.
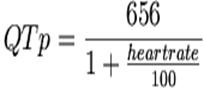
Some
great websites: www.qtsyndrome.ch